MUSCARI
Multi-task Spectral Clustering AlgoRIthm
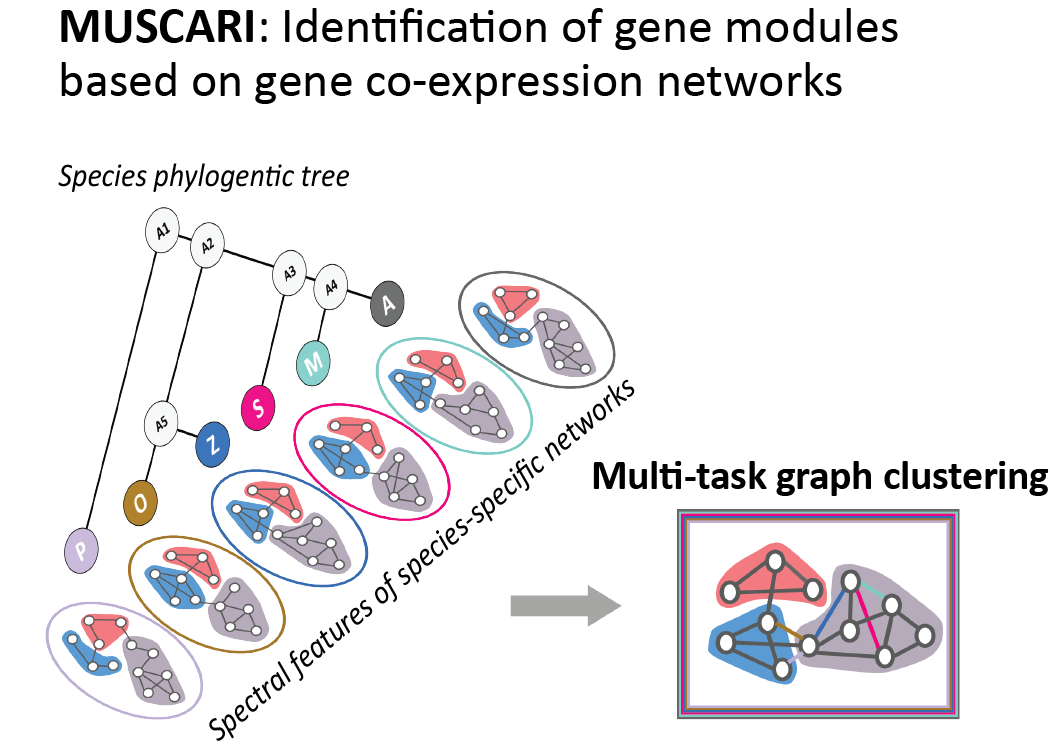
MUSCARI (Multi-task Spectral Clustering AlgoRIthm)
Muscari is a new multi-task graph-based clustering algorithm developed for the identification of gene co-expression modules (or subnetworks) jointly across species from species-specific genome-wide co-expression networks while leveraging both the phylogenetic relationship between species and the graph-based nature of co-expression matrices. Muscari is based on the Arboretum-HiC (https://doi.org/10.1186/s13059-016-0962-8) multi-task graph clustering algorithm, which defines each task as a spectral graph clustering problem, one for each species, and the the multi-task learning framework simultaneously searches for groups of genes that are interacting in multiple species while accounting for the phylogenetic relationship between species. Unlike Arboretum-HiC, Muscari inputs are gene expression matrices, which are converted into fully-connected weighted gene co-expression networks.
INSTALLATION
1. Required environment
- matlab (R2006a or higer) : required for generation of eigenvector matrices only
2. Download the code
- code/: Muscari codes directory
- run_muscari.sh : Wrapper running script for muscari. This script requires 2 other subsequent scripts:
- eigvecmat_calc.m : eigenvector matrix calculation script
- run_eigvecmat_calculation.m : wrapper script for running eigvecmat_calc.m
3. Compling Muscari code
Run code/Makefile :
makeIf the compiling was successful, you should be able to find the program named “muscari” in the code directory.
RUNNING MUSCARI
1. Preparation of input requirement files
Following is the list of files should be prepared for the running of Mucari. Please follow the description carefully.
You can also find the example files for each requirement at sample_data direcotry.
- Requirement 1: Species tree (text file)
Species tree should be prepared as a text file consist of 3 columns (tab delimited). Each row is explaining the relationship of a parent node and a child node. e.g., if species “osa” and “zma” are 2 children of ancestral node “Anc5”, this relationship could be expressed as like below:
osa (TAB) left (TAB) Anc5 zma (TAB) rigth (TAB) Anc5One parental node always have 2 children nodes (left and right). The left/right children nodes will be dealt as equivalent, i.e. there is no order between children nodes. An ancestral node could be a child of another superordinate ancestral node. e.g.
stu (TAB) left (TAB) Anc3 Anc4 (TAB) right (TAB) Anc3Refer to this file: sample_data/SpeciesTree.txt
- Requirement 2: Species order file (text file)
A simple list of extant nodes of the Species tree (requirement 1), without ancestral nodes (no “AncXX”). 1 column with only node (species) names. e.g.
ppa osa zma stu mtr athRefer to this file: sample_data/SpeciesOrder.txt
- Requirement 3: orthogroup file (text file)
A list of orthogroups(OGs) with a profiled list of corresponding gene IDs per species. This OG relationship of genes could be leant by orthogroup and gene tree learning.
- The format of this file is: “ OGID (TAB) GeneID,GeneID,GeneID,… “
(tab delimited between OGID and gene IDs, comma delimited among genes) - The order of gene IDs should be same to the Species order file (requirement 2).
- Duplicated OGs are designated as “number”. (such as OG2231, OG2232)
- Write “NONE” if there’s no species gene ID assigned to the OG.
OG474_1 (TAB) PP1S125_69V6,NONE,NONE,NONE,NONE,AT3G13810 OG474_2 (TAB) PP1S63_51V6,NONE,NONE,PGSC0003DMG400019342,MTR_2g099990,AT5G60470 OG474_3 (TAB) NONE,Os01g0242200,Zm00001d009030,PGSC0003DMG400024700,MTR_8g017210,AT5G66730 OG474_4 (TAB) NONE,NONE,Zm00001d039254,PGSC0003DMG400003372,MTR_4g059870,AT5G44160 (...)Refer to this file: sample_data/sample_OGID.txt
- The format of this file is: “ OGID (TAB) GeneID,GeneID,GeneID,… “
- Requirement 4: Value matrices (text files)
The expression value matrix for each species should be prepared separately as a tab delimited text files.
- First row of the file should be header for the following columns.
- First column should be gene IDs, which would corresponds to the gene IDs used in orthogroup file (requirement 3).
- Values of this matrix would be normalized gene expression values.
Gene SRX1795751 SRX1796285 SRX2035609 SRX2189162 SRX2484777 (...) AT1G01900 -0.031721 0.031721 -0.570001 0.238822 -0.130085 (...) AT1G01940 0.071255 -0.071255 -0.090784 -0.39764 -0.166357 (...) AT1G02180 -0.029421 0.029421 1.111816 -0.420548 -0.351622 (...) AT1G03100 -0.017806 0.017806 0.062057 0.338907 -0.028584 (...) (...)Refer to this file: sample_data/(species)_sample_matrix.txt
- Requirement 5: Preparation of config file (text file)
You need to write a simple text file for matching the species name to each eigenvector matrix and name it as “config.txt”. This file will be used another input for the clustering while the code is matching your eigenvector matrices to each species name.
- Eigenvector matrices will be named as “[species_name].eigvecs.matrix.txt” by default in our wrapper script at Step3.
- This file consists of 2 columns with tab delmited,as [species_name] (TAB) [species_name.eigvecs.matrix.txt]
ath (TAB) ath.eigvecs.matrix.txt mtr (TAB) mtr.eigvecs.matrix.txt osa (TAB) osa.eigvecs.matrix.txt ppa (TAB) ppa.eigvecs.matrix.txt stu (TAB) stu.eigvecs.matrix.txt zma (TAB) zma.eigvecs.matrix.txtRefer to this file: sample_data/config.txt
2. Running Muscari (shell script)
We are providing a wrapper shell script run_muscari.sh, which is doing (a) eigenvector matrix calculation (MATLAB) and (b) running muscari clustering (C++).
Note that the script is adjusted to run Muscari with the sample data we are providing here at sample_data directory. Therefore, if you want to just use the run_muscari.sh, please put your requirement files prepared by above into the sample_data directory first before running.
The run_muscari.sh script requires arguments below:
- K: The number of resultant modules
- P: transition probability; starting default=0.8, trying 0.5, 0.2, …
- X: fixed covariance value; starting default=0.1, trying 0.15, 0.2, …
- best species: One of the species name which is most well-studied. The gene ID of this species will be represented instead of OGID if there is the gene in that orthogroup.
- output dir name: The name of the directory which the results will be in
USAGE: ./run_muscari.sh [K] [P] [X] [best] [output] e.g. ./run_muscari.sh 10 0.8 0.1 ath result_k10
(optional) 2-1. preparation of eigenvector matrices (MATLAB)
This optional step is a part of run_muscari.sh. which could be run automatically by run_muscari.sh but demostrating here what happens by the script.
The matlab script eigvecmat_calc.m will generate a eigenvector matrix with the user-specified k number. Input arguments are:
- K: The number of resultant module. Note that this number will be same as the number of eigenvectors. For example, if k=10, the eigenvector matrix (which is the result of the eigvecmat_cal.m script) will consist of gene vectors of 10 eigenvectors.
- output prefix: the name of output prefix (usually the species name) eigenvector matrix file.
USAGE: matlab -r eigvecmat_calc\(\'[value_matrix]\',K,\'[output_prefix]\'\) e.g. matalb -r eigvecmat_calc\(\'sample_data/ath_sample_matrix.txt\',10,\'ath\'\)The script will generate output files named: “[output_prefix].eigvecs.matrix.txt”. Please also refer to the wrapper script about the usage.
matlab -r run_eigvecmat_calculation\(10\) (k=10 for example)
(optional) 2-2. detailed parameters usages of the muscari
This running of muscari step is a part of run_muscari.sh. which could be run automatically by run_muscari.sh but demostrating here what are the parameters are.
The argument keys of the program muscari are like below:- -s: species order file name (requirement 2) - -e: orthogroup ID (OGID) to gene ID file (requirement 3) - -k: number of resultan modules - -t: species tree file name (requirement 1) - -c: config file name (made in step1) - -r: species tree file name - -o: output directory name - -m: defines the mode in which the Arboretum algorithm is to be used; learn|generate|visualize|crossvalidate are the options, default=learn - -b: most well-known studied species name - -i: initialization method for transition probabilities for cluster membership across species; uniform|branchlength, default=uniform - -p: transition proability value - -x: fixed covariation value - -w: an true|false option for writing over the results on the existing directory. default=true - -f: an true|false option for running initial clustering generation. default=true
3. Outputs
If the running of Muscari was successfully finished, you could find that bunch of result files are in the result directory you’ve specified.
The following result files are containing the most relevant information of the result of clustering:
- Output 1: [species]__speciesspecnames_clusterassign.txt (tab delimited)
These files contain the result of cluster assaignments with gene IDs, formatted as “gene ID (TAB) cluster ID”.
ath_speciesspecnames_clusterassign.txt mtr_speciesspecnames_clusterassign.txt (...) - Output 2: allspecies_clusterassign_lca_brk.txt (tab delimited)
This file contains the same cluster assignment result but it provides in a table,
where the rows are orthogroup ID or gene ID of the “best” species which was specified by user
and columns are all nodes in the species tree, i.e. all extant and acestral nodes.
This file has specific aspects in the table:- Meaning of the element values: each element vlaue of the table corresponds to the cluster assignment of each species (column) and ancestor. Negative values are exhibiting “missing” elements with reasoning, where “-1” means “missing of expression value” and “-2” means “missing of gene in the phylogeny (i.e. no ortholog gene in the gene tree)”.
- Ordering of the table and the “Dummy” lines: the table is sorted by the cluster assignment of Ancestor1 (root ancestor of the tree; positioned at the last column of the table). Dummy lines are the separators of different cluster assignments between two group of Ancestor1 clusters.